 daddydoctorgym.com il sito dove trovare alcuni consigli per il buon uso della palestra e la donazione di sangue
daddydoctorgym.com il sito dove trovare alcuni consigli per il buon uso della palestra e la donazione di sangue
Nel 1973 Paul Brazeau e Coll. hanno individuato un ormone polipeptidico di 14 aminoacidi di produzione ipotalamica e dalle cellule APUD, acronimo di Amines Precursor Uptake and Decarboxilation,

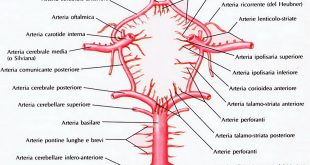
intestinali con emivita di 3-5 minuti (fonte: Rosato L. et Coll. I tumori neuroendocrini. Club delle U.E.C.; nov. 2003, pag.272); nel pancreas si ritrova nelle cellule delta, nello stomaco nelle cellule del fondo ed ancora nelle cellule intestinali. La sua presenza anche nel SNC giustifica il termine di asse cervello-intestino tramite la via ematica e la funzione del nervo vago.
Ha funzione inibitoria su:
G.H. e prolattina,
pancreas esocrino con HCl (ipocloridria, dispepsia) ed enzimi pancreatici + bicarbonato (diarrea e steatorrea con calo del peso corporeo) ed
endocrino come insulina (diabete mellito non grave e ben controllabile o RTG) e glucagone,
peptidi gastro-intestinali come gastrina, CCK, GIP, VIP, secretina, motilina,
secrezione di H2O ed elettroliti intestinali,
assorbimento di glicidi, lipidi, protidi e calcio,
attività motoria della colecisti (colelitiasi e sabbia biliare) e dell’intestino,
la differenziazione e la crescita delle cellule (GH mediato tramite l’IGF-1?), anche di quelle del sistema neuro-endocrino,
infine
< la pressione venosa del distretto mesenterico superiore e quindi del sistema portale.
Il suo impiego clinico si può avere nella profilassi (CPRE ed altre manovre endoscopiche sul duodeno-papilla di Vater) e terapia delle pancreatiti acute e nel tentativo di controllo medico delle emorragie intestinali “alte”, tipo varici esofagee sanguinanti, emorragie da ulcere o flogosi emorragiche esogago-gastro-duodenali, angiodisplasie digiunali, emorragie in corso di definizione: tutte si manifestano con melena (emissione dall’ano di feci picee, poltacee) e talora ematemesi (vomito di sangue di origine digestiva alta) ed ancora nella diagnostica in Medicina Nucleare di alcuni carcinoidi.
 Il tumore neuroendocrino ben differenziato secernente somatostatina, della famiglia degli apudomi, è il somatostatinoma, originantesi dalle cellule D del pancreas (45% ca.) e della papilla di Vater ed il duodeno (55% ca.). Nella papilla l’ittero ostruttivo intermittente-remittente, un’emorragia digestiva alta, la pancreatite, l’assenza o la modestia dei fenomeni inibitori evidenti, tipo RTG per bassa produzione dell’ormone e la possibile associazione con la malattia di Von Recklinghausen (neurofibromatosi di tipo 1) nelle sedi per lo più duodenali, governano il quadro clinico, talora molto povero in realtà. Altre volte la sintomatologia comprende la triade classica + il calo ponderale: diarrea grassa o steatorrea per mancata produzione di enzimi digestivi, diabete mellito da insulina inibita, colelitiasi colesterinica da atonia della muscolare della parete colecistica.
Il tumore neuroendocrino ben differenziato secernente somatostatina, della famiglia degli apudomi, è il somatostatinoma, originantesi dalle cellule D del pancreas (45% ca.) e della papilla di Vater ed il duodeno (55% ca.). Nella papilla l’ittero ostruttivo intermittente-remittente, un’emorragia digestiva alta, la pancreatite, l’assenza o la modestia dei fenomeni inibitori evidenti, tipo RTG per bassa produzione dell’ormone e la possibile associazione con la malattia di Von Recklinghausen (neurofibromatosi di tipo 1) nelle sedi per lo più duodenali, governano il quadro clinico, talora molto povero in realtà. Altre volte la sintomatologia comprende la triade classica + il calo ponderale: diarrea grassa o steatorrea per mancata produzione di enzimi digestivi, diabete mellito da insulina inibita, colelitiasi colesterinica da atonia della muscolare della parete colecistica.
Tumore molto raro, predilige il sesso femminile; è prevalentemente singolo, > i 3 cm. di diametro, con metastasi ai linfonodi loco-regionali ed al fegato, più tardivamente alle ossa. La diagnosi si avvale della evidenziazione immuno-istochimica nel plasma dell’ormone specifico, talora con necessità di effettuare test di stimolo con calcio, secretuina o pentagastrina. La terapia è chirurgica, spesso maggiore, laddove possibile, di pertinenza di Centri di eccellenza; talora si ferma alla biopsia per esame istologico.
foto da Sabiston 1997, T. of Surgery.

2 commenti
Pingback: GH: l’ormone della crescita. Tesi della Dr.ssa Flavia Colacioppo « daddydoctorgym.com
Pingback: Scienze Infermieristiche e Corso O.S.S. DierreForm. Patologie fisiche, lezione 33, modulo 12 - daddydoctorgym.com